The RF PeakCall module takes two RC files generated by the RF Count module, and performs transcriptome-wide peak calling from RNA immunoprecipitation (IP) experiments.
Analysis is performed by sliding a window of length w along the transcript, and calculating the signal enrichment in the IP sample versus control sample as:
where i and i+w are the start and end position of the window, μIP(i) and μCtrl(i) are respectively the coverage at position i in the IP and control samples, MdIP and MdCtrl are respectively the median coverage on the whole transcript in the IP and control samples, and p is a pseudocount added to deal with non-covered regions/transcripts.
When a control sample is not provided, the signal enrichment is simply calculated as:
A p-value is then calculated for each window with detected enrichment above a defined cutoff, using a Fisher test. Thus, the following 2x2 contingency matrix is defined for each cutoff-passing window:
| n11 | n12 | |
|---|---|---|
| n21 | μIP(i..i+w) | MdIP |
| n22 | μCtrl(i..i+w) | MdCtrl |
If no control sample is provided, the contingency matrix is instead defined as:
| n11 | n12 | |
|---|---|---|
| n21 | μIP(i..i+w) | MdIP |
| n22 | μIP windows | MdIP |
where μIP windows is the average of the mean values for each possible window in the IP sample.
P-values are then subjected to Benjamini-Hochberg correction. Consecutive significantly enriched windows are then merged together, and p-values are combined by Stouffer's method.
Since version 2.9.1, RF PeakCall can use R to generate coverage plots for individual transcripts, as well as meta-gene (a) and meta-protein-coding-gene (b) plots across the entire experiment:

Peak refinement
Peaks are called by sliding a window of a fixed size, with a user-defined offset. This means that, in some cases, the called interval might not include the full peak (or it might include additional low-enrichment bases). Consider the following example:
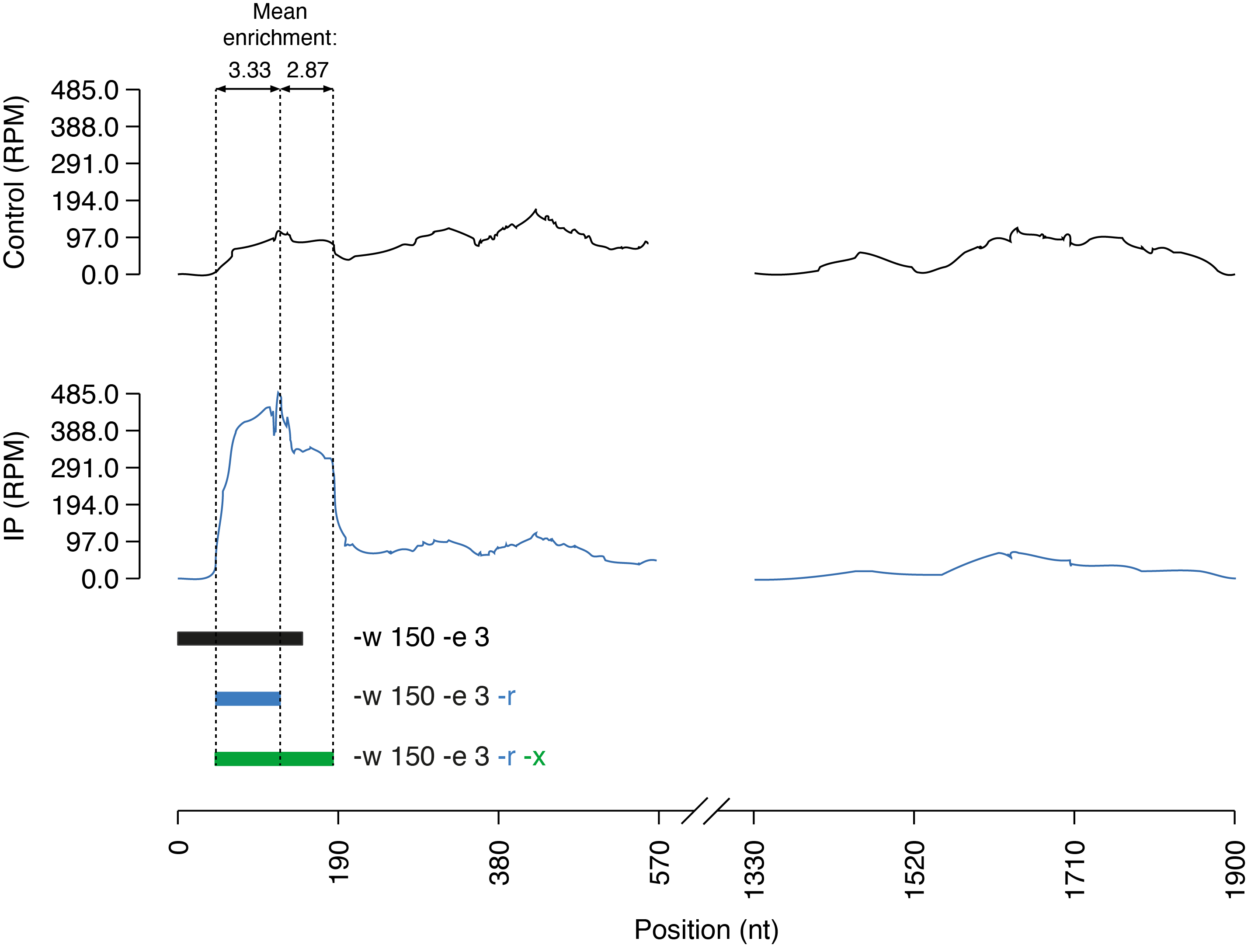
In this example, peaks are called using a window size of 150 nt (default offset: 75 nt) and an enrichment threshold of 3 fold. With default parameters, the called interval does not entangle the full peak (as the coverage slightly drops around half of the peak in the IP sample). When the -r (or --refine) parameter is specified, RF PeakCall will progressively enlarge (or shrink) the interval to only include bases exceeding the enrichment threshold. In this case, refinement trims all the non-enriched bases from the 5' end of the interval. However, because of the slightly lower coverage of the second half of the peak, the enrichment (2.87) is just below the acceptance threshold. To account for such situations, the -x (or --relaxed) parameter can be specified to enable a more loose evaluation of the coverage in the IP sample. Specifically, the coverage will be rounded to nearest multiple of 0.5.
Note
Peak refinement also takes into account the median transcript coverage of the IP sample. While extending peak boundaries, only bases exceeding the median transcript coverage in the IP sample will be included, independently of their enrichment.
Usage
$ rf-peakcall [options] -c control.rc -I IP.rc
$ rf-peakcall [options] -I IP.rc
To list all available parameters, simply type:
$ rf-peakcall -h
| Parameter | Type | Description |
|---|---|---|
| -c or --control | string | Path to the RC file for the control sample |
| -I or --IP | string | Path to the RC file for the immunoprecipitated (IP) sample |
| -i or --index | string[,string] | A comma separated (no spaces) list of RCI index files for the provided RC files Note #1: RCI files must be provided in the order 1. Control, 2. IP Note #2: If a single RTI file is specified, it will be used for all RC files Note #3: If no RCI index is provided, it will be generated at runtime, and stored in the same folder of the control/IP samples |
| l or --whitelist | string | A whitelist containing transcript IDs (one per each row) to restrict the analysis to |
| -p or --processors | int | Number of processors (threads) to use (Default: 1) |
| -o or --output | string | Output folder (Default: <IP>_vs_<Control>/ if a control RC file is provided, or <IP>/ if only the IP RC file is provided) |
| -ow or --overwrite | Overwrites the output folder (if the specified folder already exists) | |
| Peak calling options | ||
| -w or --window | int | Window size (in nt) for peak calling (≥10, Default: 150) |
| -f or --offset | int | Offset (in nt) for window sliding (≥1, Default: window / 2) |
| -md or --merge-distance | int | Maximum distance (in nt) for merging non-overlapping windows (≥0, Default: 50) |
| -e or --enrichment | float | Minimum log2 enrichment in IP vs. Control for reporting a peak (≥1, Default: 3) |
| -v or --p-value | float | P-value cutoff for reporting a peak (0 ≤ p ≤ 1, Default: 0.05) |
| -s or --summit | Generates an additional BED file containing the coordinates of peak summits (peak regions with the highest coverage) | |
| -r or --refine | Refines peak boundaries | |
| -x or --relaxed | Uses more relaxed criteria to refine peak boundaries (requires -r) |
|
| -pc or --pseudocount | float | Pseudocount added to read counts to avoid division by 0 (>0, Default: 1) |
| -mc or --mean-coverage | float | Discards any transcript with mean coverage in control sample below this threshold (≥0, Default: 0) |
| -ec or --median-coverage | float | Discards any transcript with median coverage in control sample below this threshold (≥0, Default: 0) |
| -D or --decimals | int | Number of decimals for reporting enrichment/p-value (1-10, Default: 3) |
| Plotting options | ||
| -g or --img | Enables the generation of coverage plots (one per transcript; requires R) | |
| -mp or --meta-plot | Enables the generation of meta-gene plots (requires R) | |
| -mcp or --meta-coding-plot | Enables the generation of a protein-coding-only meta-gene plot, by aligning the TSS, start codon, stop codon, and TES (requires R) | |
| -of or --orf-file | string | Path to a BED file containing the transcript-level coordinates of the CDSs Note: if no file is provided, the longest ORF will be automatically identified using the following parameters |
| -mo or --min-orf-length | int | Minimum length (in aa) to select the longest ORF (requires -mcp, Default: 50) |
| -als or --alt-start | The longest ORF is allowed to start with alternative start codons (requires -mcp) |
|
| -ans or --any-start | The longest ORF is allowed to start with any codon (requires -mcp) |
|
| -gc or --genetic-code | int | Genetic code table for the reference organism (requires -mcp, 1-33, Default: 1)Note: for a detailed list of the available genetic code tables, please refer to the RF Mutate docs, or to https://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi |
| -eo or --plot-enriched-only | Meta-gene plots will be calculated only on transcripts with a detected enrichment (requires -mp or -mcp) |
|
| -R or --R-path | string | Path to R executable (Default: assumes R is in PATH) Note: also check $RF_RPATH under Environment variables |
Output BED files
The peaks BED file contains 5 tab-delimited fields:
| Field | Description |
|---|---|
| Transcript ID | ID of the transcript |
| Start | Start coordinate of the peak (0-based) |
| End | End coordinate of the peak (1-based) |
| Enrichment | Fold enrichment of the IP signal versus Control signal |
| p-value | Benjamini-Hochberg adjusted p-value |
The summits BED file only contains 3 tab-delimited fields:
| Field | Description |
|---|---|
| Transcript ID | ID of the transcript |
| Start | Start coordinate of the summit (0-based) |
| End | End coordinate of the summit (1-based) |